おそらく、この問題を回避する最も簡単な方法は、 Bioinformatics Toolbox関数SEQLOGOのコードを直接変更することです(可能な場合)。R2010bでは、次のことができます。
edit seqlogo
そして、関数のコードがエディターに表示されます。次に、次の行(267〜284行目)を見つけて、コメントアウトするか、完全に削除します。
S_before = log2(nSymbols);
freqM(freqM == 0) = 1; % log2(1) = 0
% The uncertainty after the input at each position
S_after = -sum(log2(freqM).*freqM, 1);
if corrError
% The number of sequences correction factor
e_corr = (nSymbols -1)/(2* log(2) * numSeq);
R = S_before - (S_after + e_corr);
else
R = S_before - S_after;
end
nPos = (endPos - startPos) + 1;
for i =1:nPos
wtM(:, i) = wtM(:, i) * R(i);
end
次に、この行をその場所に配置します。
wtM = bsxfun(@times,wtM,log2(nSymbols)./sum(wtM));
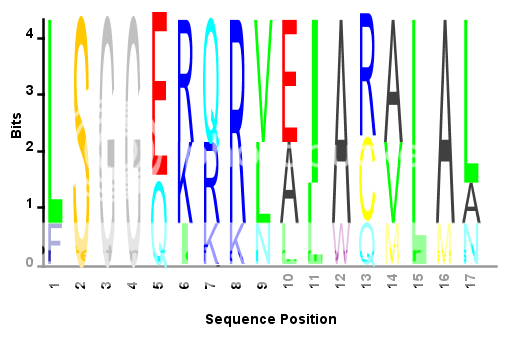
おそらく、のような新しい名前でファイルを保存することをお勧めします。そうすれば、元の変更されていないSEQLOGO関数seqlogo_norm.mを引き続き使用できます。これで、すべての列が同じ高さに正規化されたシーケンスプロファイルプロットを作成できます。例えば:
S = {'LSGGQRQRVAIARALAL',... %# Sample amino acid sequence
'LSGGEKQRVAIARALMN',...
'LSGGQIQRVLLARALAA',...
'LSGGERRRLEIACVLAL',...
'FSGGEKKKNELWQMLAL',...
'LSGGERRRLEIACVLAL'};
seqlogo_norm(S,'alphabet','aa'); %# Use the modified SEQLOGO function
古い答え:
シーケンスプロファイル情報を変換して、 Bioinformatics Toolbox関数SEQLOGOから目的の出力を取得する方法はわかりませんが、リンクした関連質問への回答のseqlogo_new.mために書いた代替案を変更する方法を紹介できます。これから初期化する行を変更した場合:bitValues
bitValues = W{2};
これに:
bitValues = bsxfun(@rdivide,W{2},sum(W{2}));
次に、各列を高さ1にスケーリングする必要があります。次に例を示します。
S = {'ATTATAGCAAACTA',... %# Sample sequence
'AACATGCCAAAGTA',...
'ATCATGCAAAAGGA'};
seqlogo_new(S); %# After applying the above modification
